Investigating the Current Limitations of the Pharma Industry — And How Personalized Medicine Can Solve Them
Diseases are on the rise. Our drug industry is faltering. But personalized medicine could solve it all.
Are we making significant progress in the area of drug discovery?
Well, about 500 drugs are released by pharma companies each year into the FDA’s public pipeline, and while larger amounts of publicly available drugs imply a larger range of diseases covered, other statistics reveal that clinical conditions worsening, both in number and degree.
“Many or most new drugs are not — or at least not provably — an improvement over the best existing drug for a given condition, and the fast-track drug-approval processes that have prevailed in recent years have added to the uncertainty about their advantages” — Claudia Wells from “A Dilemma with New Drugs” (Scientific American November 2019 Issue)

We might be reaching a dead-end in Drug Discovery. According to a recent article published in the British Medical Journal, ~50 % of drugs approved by the German Healthcare System did not help in increasing healthspan.
Consequently, the number of adverse drug reactions (ADRs), serious side-effects to a drug that almost outweighs its benefits, has doubled in the last decade.
What are we doing wrong? What’s hindering progress in this field?
In this article, we’ll explore Lipinksi’s Rule and the pharmacokinetics of drug development (how the drug is absorbed into the blood, distributed throughout the body, metabolized, and excreted (This is all encapsulated in an acronym called ADME)). As we go through topics, we’ll see how traditional practices and concepts in drug development might be constraining the field. In pharmacokinetics, we’ll see how genetic mutations that vary from person to person affect various steps in this process, and how these deviations can lead to devastating consequences.
Before continuing with reading this article, I’d highly recommend that you guys read my first article on drugs and drug development!
Lipinski’s Rule of five
Lipinski’s Rule of Five establishes relatively rigid parameters that restrict the chemical structure of a drug. When drugs are distributed throughout the body, they must pass through a variety of environments. For example, to dissolve in the blood, which is mostly made up of water, a drug must be partly hydrophilic. However, at the same time, to pass through a cell membrane (which is mostly hydrophobic and made of nonpolar phospholipids), it must be partially hydrophobic as well. Therefore, a delicate balance must be struck when designing a drug.
Similarly, a drug cannot be too big nor too small, nor form too many or too few hydrogen bonds. Listed below are the exact parameters according to Lipinski’s rule:
- Maximum Hydrogen Bond Donors (groups with a nitrogen or oxygen atom w/ a lone pair) = 5
- Maximum Hydrogen Bond Acceptors (N-H groups or O-H groups) = 5
- Maximum Molecular Weight for a substance = 500 Daltons
Lipophilicity is measured through a value called log P. The drug is dissolved in a mixture of octane and water, and the lipophilicity is calculated like this:
- log ([solute] in octane / [solute] in water)

Lipinski’s rule is extremely rigid. Although it serves as a mere framework that establishes chemical parameters for drug development, solely following these parameters can lead to a standstill in drug design and development.
Before we go on further in this article, it is important that we define two key terms:
- Potency: Potency is measured by the amount of drug required to produce a given response
- Efficacy: Efficacy is measured by the maximal response that a drug can produce

ADME (Absorb, Distribute, Metabolize, Excrete)
Our bodies are like factories and manufacturing lines that take in recycled parts. First, the parts are taken into the factory, then they are distributed across the factory and used for whatever purposed they were obtained for. Finally, any remaining parts or waste products are broken down and sent elsewhere.
In the same way, our bodies take in drugs, use them for a purpose, metabolize the drugs, and then excrete them. Although the binding of the ligand to the receptor is the main step that sets off a biochemical cascade, all the other parts of the process as equally as important in determining the potency and side effects of the drug.
In the section below, I’ll cover the two main parts of ADME that serve as the main source of variation from person to person: Absorption and Metabolism.
Absorption
When a drug is absorbed into the bloodstream, it must first be absorbed into the blood plasma, the part containing water, electrolyte, or other proteins. Now, placing the drug in the circulatory system serves as a great mechanism for distribution; however, the drug may also interact with other proteins that are in the blood plasma. In fact, almost 8% by weight of the blood serum is made up of protein… and now we have another complication that varies from person to person.😔
Albumin constitutes a majority of the blood serum and potentially binds to basic ligands. Similarly, Globulins often bind basic ligands. (Here’s a super cool article that talks about how mutations (loss-of-function and gain-of-function) in the albumin gene can lead to varying concentrations of certain peptide hormones)

Metabolism
After the drug has been distributed throughout the body, the liver breaks active drugs into inactive metabolites. This step is extremely important. Let’s took at a drug called warfarin, a drug that used to prevent blood coagulation/clotting.
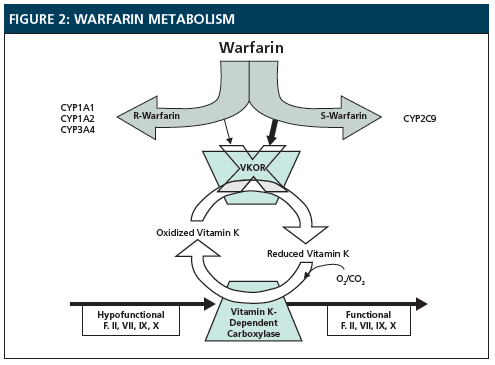
Warfarin acts by inhibiting a protein called VKOR which aids in the reduction of Vitamin K. The reduced Vitamin K then goes to aid in blood clotting. By inhibiting VKOR, Warfarin obstructs the reduction of Vitamin K, thereby preventing blood from coagulating.
Warfarin is then metabolized by the hepatic cytochrome p450s, a group of enzymes in the liver that metabolizes drugs. However, people can often have mutations in these drug-metabolizing enzymes. If someone taking Warfarin as an anti-coagulant has a mutation in the enzyme CYP2C9, then warfarin will build up in their system and can induce toxic side effects, including internal bleeding and hemorrhage.
TL;DR In order to sidestep ADRs due to mutations in drug-metabolizing enzymes, we must personalize drug dosage for every individual.
Target Vs. Phenotype Based Drug Discovery
In ancient times, physicians obviously not try and find drugs that bound to a particular target. Rather, they used trial and error to find herbs that cured the symptoms for a particular phenotype. Therefore, all the elements of ADME would inherently be inculcated into the given cure, and side effects would automatically be reduced.
Nevertheless, there are some downsides to phenotype-based drug discovery. Since the efficacy of a drug is determined by outward symptoms, the protein target of a drug is not known. Not knowing the target could have a variety of unforeseen consequences.
Target deconvolution is the process by which scientists work backward to identify which protein was affected by a drug. It sounds easy but it really isn’t. Scientists have to act like detectives, searching hundreds of relevant proteins in the cell to find the correct one.
Key Takeaways:
- The current process for drug design and development is a lengthy process, costing billions of dollars and decades of work, but we aren’t seeing the fruits of this effort. We need to consider personalizing certain aspects of the process such as absorption and metabolism to make progress in this industry and avoid ADRs.
- Lipinski’s Rule of Five dictates certain parameters for a drug that governs its lipophilicity and size, but these parameters are rigid, and we need to think more critically. What if we modified Lipinski’s rule based on certain classes of mutations in blood serum proteins? This could be a possible way to avoid some of the ADRs caused due to a lack of bioavailability.
- Target-based drug design has been the focus of the past few decades…but what if we started to pay more attention to phenotype-based drug discovery to discover more personalized drugs? Target deconvolution might be a lengthy and challenging process, but techniques such as RNAi (RNA interference), cell-based biochemical assays, and ML algorithms might help us with this.
Thanks for reading! Feel free to check out my other article on Medium and connect with me on LinkedIn!
If you’d like to discuss any of the topics above, I’d love to get in touch with you!
Please sign up for my monthly newsletter here if you’re interested in following my progress :)
